La purificación de proteínas es el proceso de introducir secuencias de ácidos nucleicos que codifican proteínas en las células huésped mediante técnicas de ingeniería genética, haciendo que se expresen en grandes cantidades y luego purificándolas in vitro utilizando métodos de purificación adecuados para obtener proteínas de alta pureza, actividad y rendimiento . Los sistemas de expresión de proteínas se pueden clasificar en sistemas procarióticos y sistemas eucariotas..
los principios y métodos para inducir la expresión de proteínas en sistemas procarióticos y purificar proteínas in vitro mediante cromatografía de afinidad . A continuación se describen
PRINCIPIO
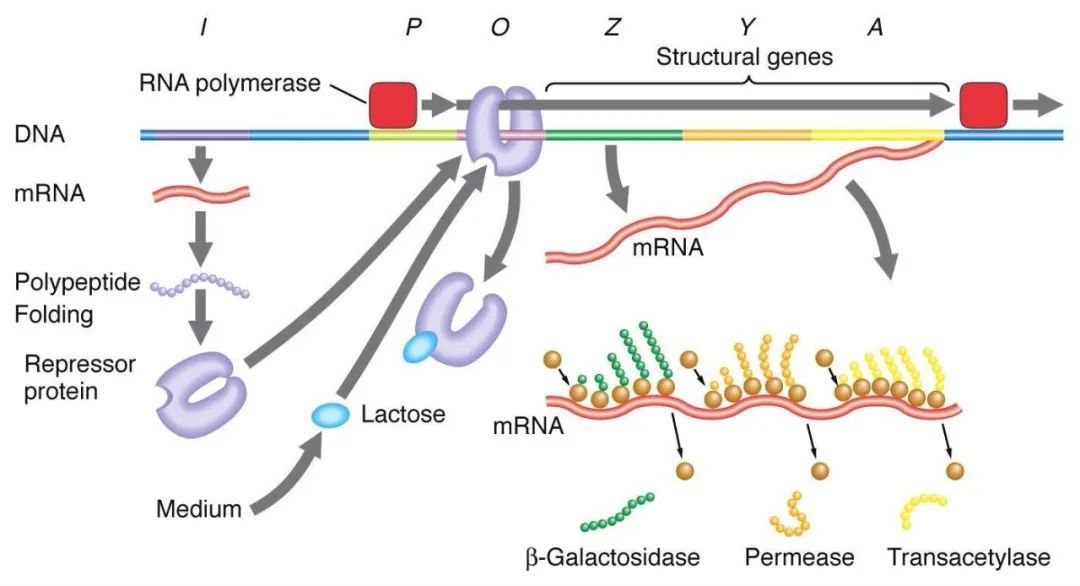
1. Regulación negativa del manipulador de lactosa:
En ausencia de lactosa, el manipulador lac se encuentra en un estado de disuasión, momento en el que la secuencia I expresa la proteína disuasoria Lac para unirse a la secuencia O, evitando que la ARN polimerasa se una a la secuencia P e inhibiendo el inicio de la transcripción. Cuando hay lactosa presente, la lactosa ingresa a la célula y es catalizada por la β-galactosidasa y se convierte en isolactosa, que se une a la proteína inhibidora, lo que provoca un cambio conformacional en la proteína y conduce a la disociación de la proteína inhibidora de la secuencia O, iniciando así la transcripción.
El isopropiltiogalactósido (IPTG) actúa de la misma forma que la isogalactosa y es un inductor extremadamente potente que no es metabolizado por bacterias y es muy estable, por lo que se utiliza mucho en los laboratorios.
2 、 selección de cepa huésped:
(1) BL21 es una de las cepas más utilizadas para la expresión procariótica, que es principalmente adecuada para la expresión de proteínas no tóxicas con polimerasa de E. coli, por lo que se puede aplicar a la expresión de sistemas procarióticos utilizando ARN polimerasa de E. coli como tac o trc (por ejemplo, plásmidos pGEX, pMAL).
(2) BL21(DE3) integra el gen de la ARN polimerasa del fago T7 en la región DE3 del fago λ en el cromosoma de la cepa BL21, que puede expresar tanto la ARN polimerasa T7 como la ARN polimerasa de E. coli , y puede usarse para la expresión de plásmidos como la serie pET, pGEX, pMAL, etc.
3, métodos de purificación de proteínas:
(1) Cromatografía de filtración en gel: de acuerdo con el tamaño molecular de la mezcla para separar proteínas, la forma de diferentes proteínas y el tamaño molecular de las diferencias en la mezcla a través de la columna de cromatografía de filtración en gel que contiene partículas de relleno, debido al tamaño molecular de varias proteínas son diferentes, la difusión en el tamaño específico de las partículas de apertura de la capacidad de variar, cuanto más grandes sean las moléculas de proteína, las primeras en ser eluidas de las moléculas son más pequeñas cuanto más tarde, más tardía es la elución.
(2) Cromatografía de intercambio iónico: la separación y purificación de proteínas se basa en las diferentes cargas en la superficie de la proteína, la superficie de la proteína generalmente está cargada uniformemente y se puede combinar con columnas de intercambio catiónico/aniónico bajo ciertas condiciones. Cuando se cambia el pH o se utiliza para la elución un tampón con fuerza iónica que aumenta gradualmente, la sustancia unida se puede intercambiar con los iones del eluyente y eluir en la solución. Dado que diferentes sustancias tienen diferentes cargas y diferentes capacidades de unión con la columna de intercambio iónico, el orden de elución en la solución también es diferente.
(3) Cromatografía hidrofóbica: al utilizar la hidrofobicidad de las proteínas, los residuos hidrofóbicos quedarán expuestos en la superficie de las proteínas después de la desnaturalización o en un ambiente con alto contenido de sal, los residuos hidrofóbicos de diferentes proteínas tienen diferentes fuerzas de acción con los ligandos hidrofóbicos de la fase estacionaria, y la hidrofobicidad se puede utilizar como una separación de componentes de más débil a más fuerte utilizando la fuerza iónica del eluyente en orden de mayor a menor.
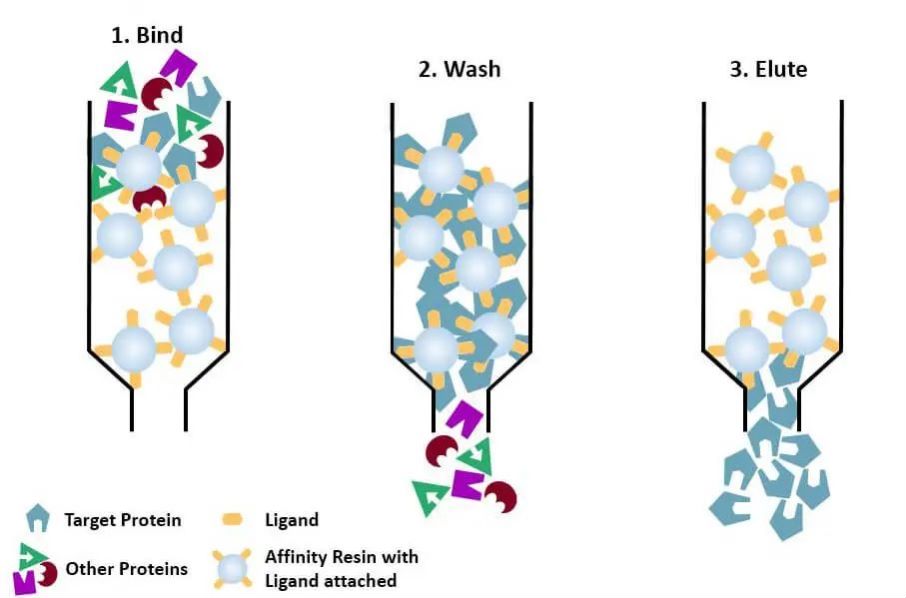
(4) Cromatografía de afinidad: el ligando específico de una proteína que se va a purificar (o etiquetar en la proteína) se une covalentemente a la molécula portadora mediante métodos químicos apropiados. Cuando la mezcla de proteínas se añade a la columna cromatográfica llena de medio de afinidad, la proteína que se va a purificar se une específicamente al ligando, mientras que las otras proteínas no se unen ni se eliminan mediante lavado, y la proteína unida específicamente se puede eluir con una solución del ligando correspondiente libre. Las proteínas unidas específicamente se pueden eluir con una solución que contenga el ligando correspondiente libre.
4 、 Selección de etiquetas de purificación de proteínas:
Las características de las diferentes etiquetas son las siguientes↓
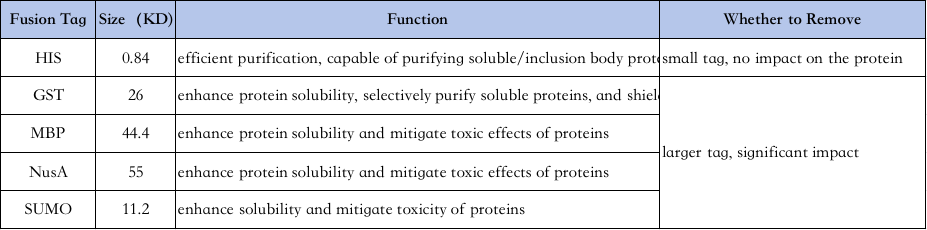
Purificación de etiqueta His: La etiqueta His es una de las etiquetas más comúnmente utilizadas, donde se agregan de 6 a 10 histidinas al extremo amino o carboxilo de la proteína, y la proteína objetivo se purifica por su capacidad para unirse firmemente a columnas quelantes de Ni2+ en condiciones normales o desnaturalizantes (p. ej., urea 8 M) y luego se eluye con imidazol (que compite por la unión a iones de níquel en los pocillos de perlas).
Instrumentos y consumibles
Reactivos de laboratorio:
IPTG; medio LB;
Tampón de lisis: Tris 20 mM (7,9), NaCl 500 mM, imidazol 10 mM;
Tampón de lavado: Tris 20 mM (7,9), NaCl 500 mM, imidazol 20 mM;
Tampón de elución: Tris 20 mM (7,9), NaCl 200 mM, imidazol 300 mM ( la concentración de imidazol se puede ajustar según la eficacia de elución );
Solución de diálisis: Tris 20 mM (7,9), NaCl 50 mM, glicerol al 10%;
Solución de tinción Kaomas Brilliant Blue; solución decolorante
Instrumentos:
Ni-NTA, disyuntor ultrasónico, columna de cromatografía, tubos de concentración

Procedimiento
1. Construcción de plásmido de expresión procariota: PCR del gen diana, digestión del vector, ligación, transformación, selección de un solo clon para secuenciación;
2. Transformar el plásmido construido con éxito en BL21(DE3), incubar a 37 ℃ durante la noche y seleccionar el clon único para agitarlo ligeramente durante la noche;
3 、 Pequeña inducción para descubrir las mejores condiciones de inducción: se agitará durante la noche la solución bacteriana 1:1000, se inoculará en 3 ml de LB, se agitará a 37 ℃ durante 4-5 h para la solución bacteriana OD600 = 0,6-0,8, se agregarán diferentes concentraciones de IPTG (0,1-1 mm), diferentes temperaturas, con la disminución de la temperatura, el tiempo de inducción se extiende, como por ejemplo a 37 ℃ inducido 4-5 h, 30 ℃ inducido 6 h-8 h, 16 ℃ inducido 16-20 h, 16 ° C inducido 16-20 h, la temperatura se reduce, el tiempo se extiende, como 37 ℃ inducido 4-5 h, 30 ℃ inducido 6 h-8 h, 16 ° C inducido 16-20 h, la temperatura se reduce. C durante 16-20 h, tomar la savia bacteriana antes y después de la inducción en diferentes condiciones de inducción, ejecutar el gel, teñir y observar los resultados de la inducción; (En general, cuanto menor sea la concentración de IPTG y menor la temperatura de inducción, más lenta será la expresión de la proteína diana, más propicia para el correcto plegamiento de las proteínas, aumentando así su solubilidad y reduciendo la generación de cuerpos de inclusión)
4. Después de encontrar las condiciones de inducción más adecuadas, inocular la solución bacteriana 1:1000 en 2 litros de LB, agitar a 37 ℃ hasta que la OD600 de la solución bacteriana = 0,6-0,8, aspirar 20 μL de la solución bacteriana y dejar correr el gel (1), luego inducir la expresión de proteínas en las condiciones de inducción adecuadas obtenidas en el experimento previo, aspirar 20 μL de la solución bacteriana y dejar. para ejecutar el gel (2);
5. Retire el líquido bacteriano inducido, centrifugue a 4000 g durante 15 minutos a 4 ℃;
6. Deseche el sobrenadante, pese, agregue 10 ml de tampón de lisis (1:100 más inhibidor de proteasa) por cada gramo de bacteria, resuspenda y coloque en hielo durante aproximadamente 30 minutos;
7. Trituración a presión: retire el alcohol en el homogeneizador de alta presión, enjuague con agua dos veces, use tampón de lisis para equilibrar una vez, agregue el líquido bacteriano, presurización (la presión no debe exceder los 800 kpa), el líquido bacteriano de tres a cinco veces hasta que sea transparente y no pegajoso;
8. Recoger el líquido bacteriano triturado, 12000 g, centrifugar a 4 ℃ durante 20 minutos, separar el sobrenadante y el precipitado, cada uno retuvo 20 μl de muestra (3) (4) para procesar el gel;
Se agregaron 9.2 ml de Ni-NTA a la columna de purificación, se filtró con etanol, se enjuagó con agua y se agregó tampón de lisis para equilibrar la columna;
10. Con la resuspensión del tampón de lisis Ni-NTA agregada al sobrenadante, mezcle bien, incubación en agitador a 4 ℃ durante 2 h;
11. Se pasó el sobrenadante a través de la columna a 4°C y se recogieron 20 µL de filtrado (5);
12. Lave la columna con 5 ml de tampón de lavado 3 veces, recoja 20 μl de muestra de filtrado (6);
13. Agregue 1 ml de tampón de elución, incube durante 5 minutos, recoja el eluido, repita 5 veces, recoja 5 ml de eluido en el mismo tubo, deje 20 μl de muestra (7);
14. Las muestras de proteínas individuales obtenidas en el transcurso del experimento se procesaron, se tiñeron con Caumas Brilliant Blue durante 1 hora y se decoloraron hasta que el color azul del fondo fue más claro y se hicieron visibles bandas de proteína claras;
15. Analice las bandas de proteínas de cada muestra, de acuerdo con los resultados de la operación posterior: si las muestras de proteínas eluidas en la proteína sin bandas heterocigotas o bandas heterocigotas o menos y poco profundas, pueden ser diálisis y concentración, si es necesario purificar más bandas heterocigotas nuevamente después de la diálisis y la concentración.
16. Diálisis: agregue la muestra a la membrana de diálisis, sujete ambos extremos, diálisis a 4 ℃ durante la noche;
17 、 Concentración: de acuerdo con el tamaño del peso molecular de la muestra para elegir el tubo de concentración apropiado, concentración a baja velocidad de 4 ℃, medición de la concentración de proteína, etiquetado, congelación rápida en nitrógeno líquido y luego congelación en un refrigerador de -80 ℃.
RESULTADO
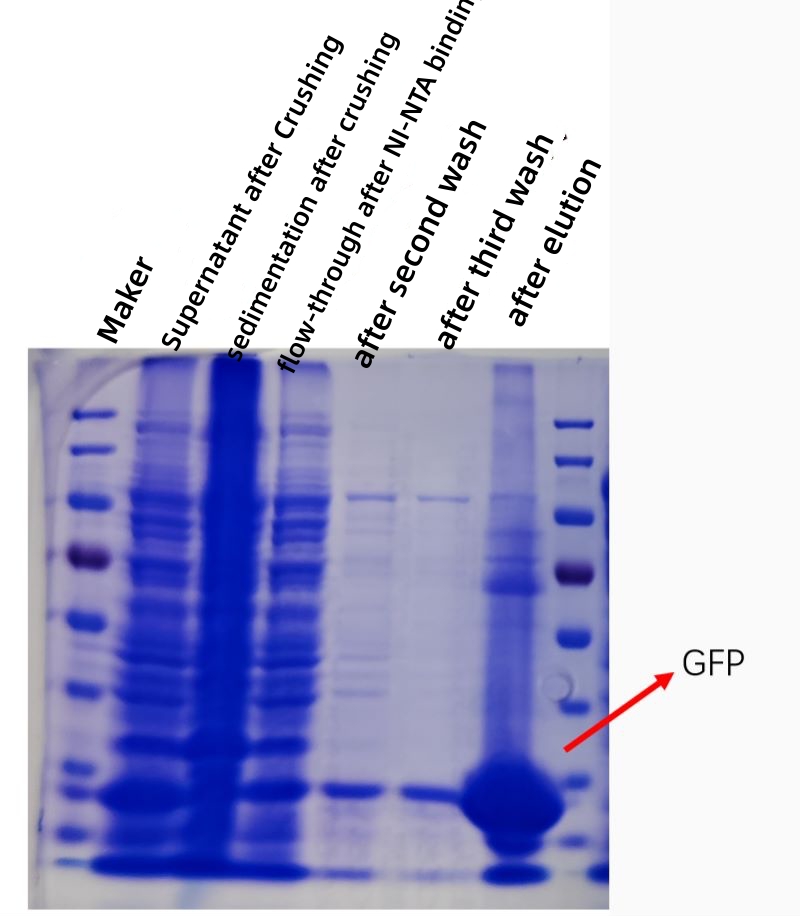
La siguiente figura muestra un gráfico de kofección de la purificación por afinidad de la proteína GFP in vitro, y la pureza y concentración de la proteína purificada aumentan considerablemente.