Protein Purification is the process of introducing nucleic acid sequences coding for proteins into host cells through genetic engineering techniques, causing them to be expressed in large quantities, and then purifying them in vitro using appropriate purification methods in order to obtain proteins of high purity, activity, and yield. Protein expression systems can be categorized into prokaryotic systems and eukaryotic systems.
The principles and methods for inducing protein expression in prokaryotic systems and purifying proteins in vitro using affinity chromatography are described below.
PRINCIPLE
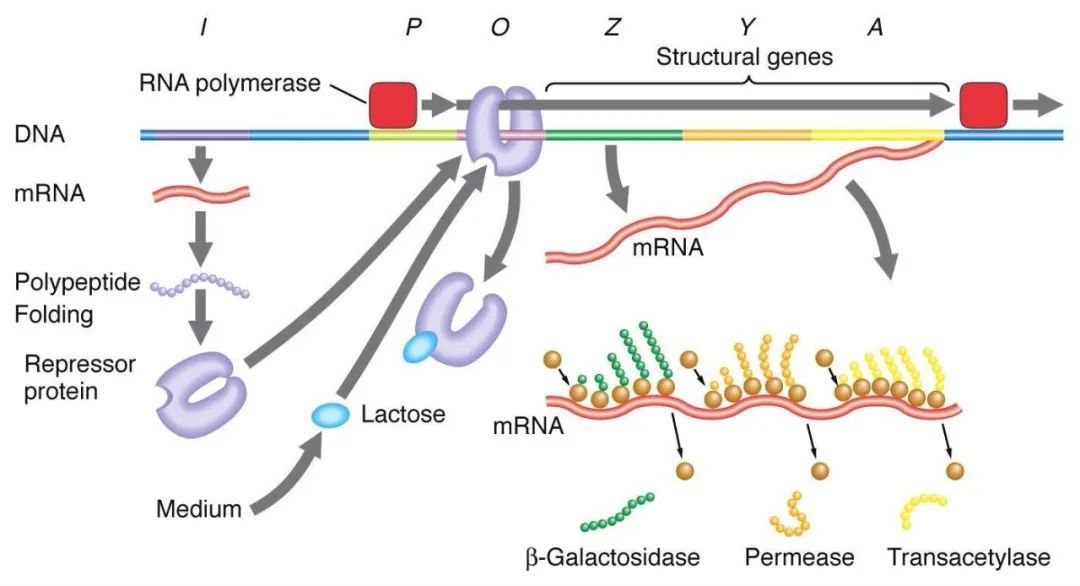
1、Negative regulation of the lactose manipulator:
In the absence of lactose, the lac manipulator is in a state of deterrence, at which time the I sequence expresses the Lac deterrent protein to bind to the O sequence, preventing RNA polymerase from binding to the P sequence and inhibiting transcription initiation. When lactose is present, lactose enters the cell and is catalyzed by β-galactosidase and converted to isolactose, which binds to the inhibitory protein, causing a conformational change in the protein and leading to dissociation of the inhibitory protein from the O sequence, thereby initiating transcription.
Isopropylthiogalactoside (IPTG) acts in the same way as isogalactose and is an extremely potent inducer that is not metabolized by bacteria and is very stable, and is therefore widely used in laboratories.
2、host strain selection:
(1) BL21 is one of the most commonly used strains for prokaryotic expression, which is mainly suitable for the expression of non-toxic proteins with E. coli polymerase, so it can be applied to the expression of prokaryotic systems using E. coli RNA polymerase such as tac or trc (e.g. pGEX, pMAL plasmids).
(2) BL21(DE3) integrates the T7 phage RNA polymerase gene in the λ phage DE3 region on the chromosome of BL21 strain, which can express both T7 RNA polymerase and E. coli RNA polymerase, and can be used for the expression of plasmids such as the pET series, pGEX, pMAL, and so on.
3, protein purification methods:
(1) Gel filtration chromatography: according to the molecular size from the mixture to separate proteins, the shape of different proteins and the molecular size of the differences in the mixture through the gel filtration chromatography column containing filler particles, due to the molecular size of various proteins are different, the diffusion into the specific size of the aperture particles of the ability to vary, the larger the protein molecules will be the first to be eluted out of the molecules are smaller the later the later the elution.
(2) Ion exchange chromatography: protein separation and purification is based on the different charges on the surface of the protein, the surface of the protein is usually uniformly charged, and can be combined with cation/anion exchange columns under certain conditions. When the pH is changed or a buffer with gradually increasing ionic strength is used for elution, the bound substance can be exchanged with the ions in the eluent and eluted into the solution. Since different substances have different charges and different binding abilities with the ion exchange column, the order of being eluted into solution is also different.
(3) Hydrophobic Chromatography: Using the hydrophobicity of proteins, hydrophobic residues will be exposed on the surface of proteins after denaturation or in a high salt environment, the hydrophobic residues of different proteins have different strengths of action with the hydrophobic ligands of the stationary phase, and hydrophobicity can be used as a weakest to strongest component separation by using the ionic strength of the eluent in order from high to low.
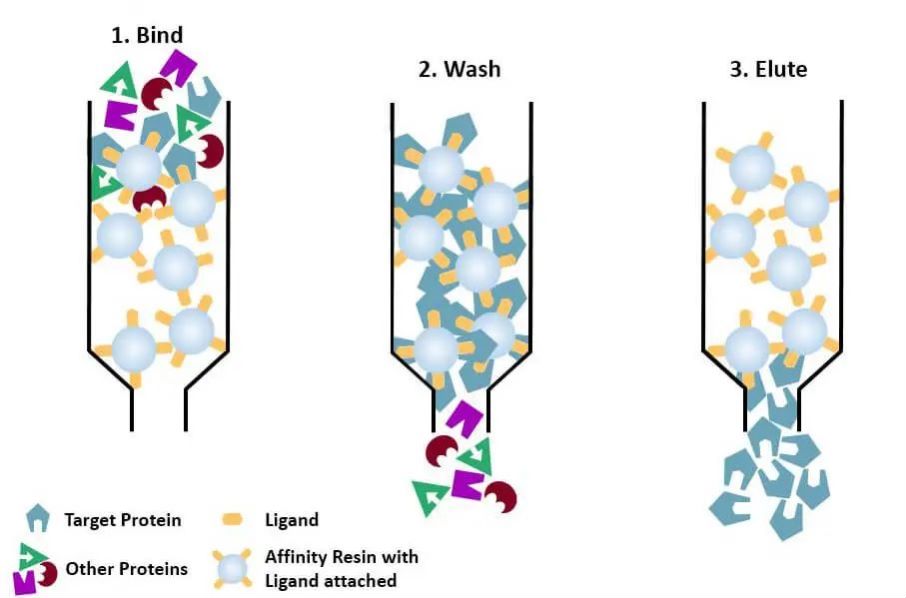
(4) Affinity chromatography: the specific ligand of a protein to be purified (or tagged on the protein) is covalently attached to the carrier molecule by appropriate chemical methods. When the protein mixture is added to the chromatographic column filled with affinity medium, the protein to be purified is specifically bound to the ligand, while the other proteins are not bound and removed by washing, and the specifically bound protein can be eluted off with a solution of free corresponding ligand. The specifically bound proteins can be eluted with a solution containing the free corresponding ligand.
4、Selection of protein purification labels:
The characteristics of different labels are as follows↓
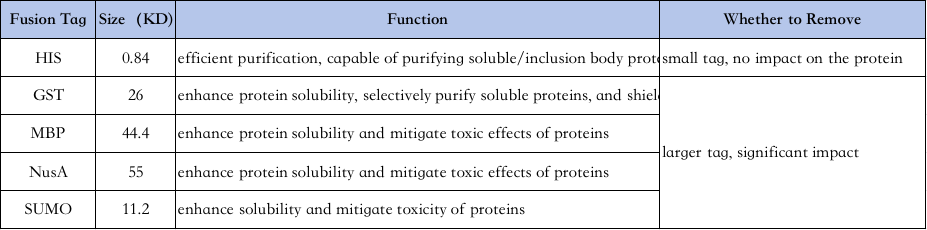
His-tag purification: His-tag is one of the most commonly used tags, where 6-10 histidine are added to the amino or carboxyl terminus of the protein, and the target protein is purified by its ability to bind tightly to Ni2+ chelating columns under normal or denaturing conditions (e.g., 8M urea), and then eluted with imidazole (which competes for binding to nickel ions in the bead wells).
Instruments and Consumables
Laboratory reagents:
IPTG; LB medium;
Lysis buffer: 20 mM Tris (7.9), 500 mM NaCl, 10 mM Imidazole;
Wash buffer: 20 mM Tris (7.9), 500 mM NaCl, 20 mM Imidazole;
Elution buffer: 20mM Tris (7.9), 200mM NaCl, 300mM Imidazole (Imidazole concentration can be adjusted according to the elution efficiency);
Dialysis solution: 20 mM Tris (7.9), 50 mM NaCl, 10% glycerol;
Kaomas Brilliant Blue staining solution; decolorizing solution
Instruments:
Ni-NTA, ultrasonic breaker, chromatography column, concentration tubes

Procedure
1、Construction of prokaryotic expression plasmid: PCR of target gene, vector digestion, ligation, transformation, picking single clone for sequencing;
2、Transform the successfully constructed plasmid into BL21(DE3), incubate at 37℃ overnight, and pick the single clone for small shaking overnight;
3、Small induction to find out the best induction conditions: will be shaken overnight bacterial solution 1:1000 inoculation into 3mL LB, 37 ℃ shaking 4-5h to the bacterial solution OD600 = 0.6-0.8, add different concentrations of IPTG (0.1-1mM), different temperatures, with the lowering of the temperature, induction time is extended, such as 37 ℃ induced 4-5h, 30 ℃ induced 6h-8h, 16 ℃ induced 16-20h, 16 ° C induced 16-20h, the temperature is reduced, the time is extended, such as 37 ℃ induced 4-5h, 30 ℃ induced 6h-8h, 16 ° C induced 16-20h, the temperature is reduced. C for 16-20h, take the bacterial sap before and after induction under different induction conditions, run the gel, stain and observe the results of induction; (Generally speaking, the lower the concentration of IPTG and the lower the induction temperature, the slower the expression of the target protein, the more conducive to the correct folding of proteins, thereby increasing their solubility and reducing the generation of inclusion bodies)
4、After finding the most suitable induction conditions, inoculate the bacterial solution 1:1000 into 2L of LB, shake at 37℃ until the OD600 of the bacterial solution=0.6-0.8, aspirate 20μL of the bacterial solution and leave it to run the gel (1), then induce the protein expression under the suitable induction conditions obtained in the pre-experiment, and aspirate 20μL of the bacterial solution and leave it to run the gel (2);
5、Remove the induced bacterial liquid, centrifuge at 4000g for 15min at 4℃;
6、Discard the supernatant, weigh, add 10mL Lysis buffer (1:100 plus protease inhibitor) for each gram of bacteria, resuspend, place on ice for about 30min;
7、Pressure crushing: put off the alcohol in the high-pressure homogenizer, rinse with water twice, use Lysis buffer to balance once, add the bacterial liquid, pressurization (pressure should not exceed 800kpa), the bacterial liquid over three to five times to transparent and not sticky;
8、Collect the crushed bacterial liquid, 12000g, 4 ℃ centrifugation for 20min, the supernatant and precipitate separation, each retained 20μL sample (3) (4) to be run gel;
9、2mL Ni-NTA was added to the purification column, to be ethanol filtered, rinsed with water, and added Lysis buffer to equilibrate the column;
10、With Lysis buffer Ni-NTA resuspension added to the supernatant, mix well, 4 ℃ shaker incubation for 2h;
11、The supernatant was passed through the column at 4°C, and 20 μL of filtrate was collected (5);
12、Wash the column with 5mL Wash buffer 3 times, collect the filtrate sample 20μL (6);
13、Add 1mL Elution buffer, incubate for 5min, collect the eluate, repeat 5 times, collect 5mL of eluate in the same tube, leave 20μL of sample (7);
14、The individual protein samples obtained in the course of the experiment were run, stained with Caumas Brilliant Blue for 1h, and decolorized until the background blue color was lighter and clear protein bands were visible;
15、Analyze the protein bands of each sample, according to the results of the subsequent operation: if the eluted protein samples in the protein without heterozygous bands or heterozygous bands or less and shallow, can be dialysis and concentration, if more heterozygous bands need to be purified again after dialysis and concentration.
16、Dialysis: add the sample into the dialysis membrane, clamp both ends, dialysis at 4℃ overnight;
17、Concentration: according to the molecular weight size of the sample to choose the appropriate concentration tube 4 ℃ low-speed concentration, concentration of protein concentration measurement, labeling, in liquid nitrogen quick-freezing, and then frozen in -80 ℃ refrigerator.
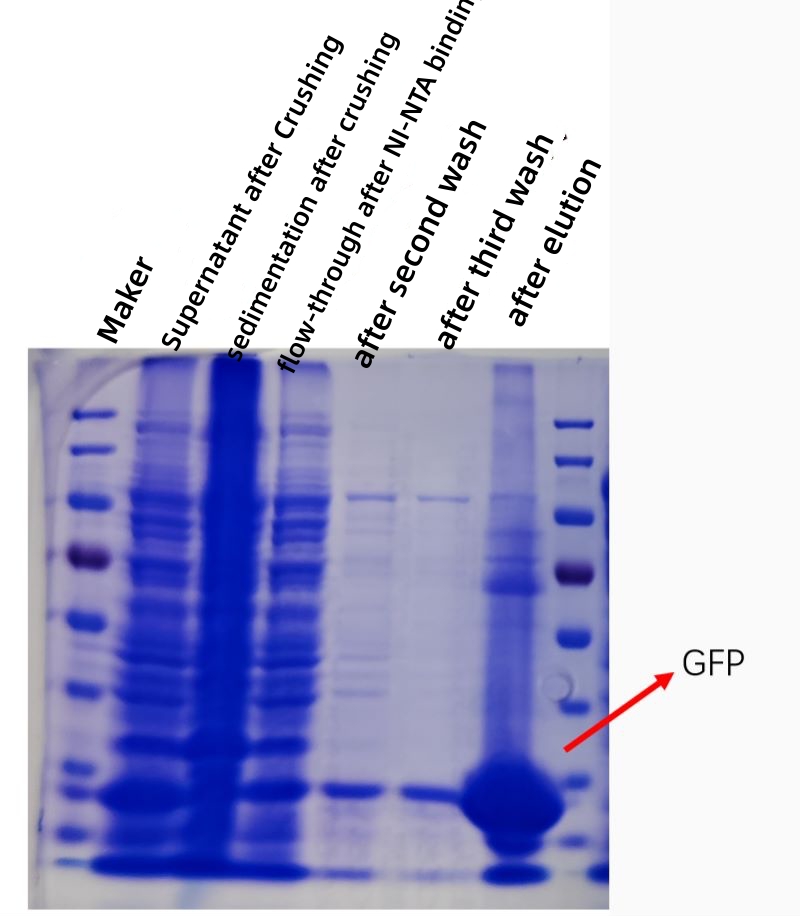
RESULT
The figure below shows a kofection plot of GFP protein affinity purification in vitro, and the purity and concentration of the purified protein is greatly increased